
We will concentrate our efforts in funding research efforts in the following 4 areas:
- Utrophin Upregulation
- Exon Skipping
- Gene Therapy: Micro-Dystrophin 4500 bases
- Gene Therapy: AAV.Mini-Dystrophin Overlapping Vectors 6400 bases
Utrophin

Muscular dystrophy is a group of diseases that cause progressive weakness and loss of muscle mass. In muscular dystrophy, abnormal genes (mutations) interfere with the production of proteins needed to form healthy muscle.
Duchenne (DMD) and Becker (BMD) muscular dystrophies are being caused by a mutation of a gene located on the X chromosome: the dystrophin Gene. Dystrophin is a member of the spectrin superfamily of proteins and is closely related in sequence similarity and functional motifs to three proteins that constitute the dystrophin related protein family, including the autosomal homologue, utrophin.
Utrophin was found during research into Duchenne’s muscular dystrophy. The name is a contraction for ubiquitous dystrophin. The 900 kb gene for utrophin is found on the long arm of human chromosome 6.
Utrophin can functionally substitute for dystrophin. However, because utrophin exists in very low levels in muscles, upregulation (increasing the production) of utrophin protein is required so that utrophin exists in muscles at levels that can compensate for lack of dystrophin.
Although there have been ongoing efforts to upregulating utrophin, so far none has proven successful. What is extremely important about using utrophin as cure for BMD and DMD is that, if successful, it will cure all phenotypes of the two diseases regardless of the mutation type or location. Essential it can cure all patients with BMD or DMD.
Exon Skipping

Exon skipping is one of the most promising therapies for Duchenne Muscular Dystrophy (DMD) today. The main goal of exon skipping therapy is to increase dystrophin levels to 10 to 25% of normal levels by restoring the expression of the mutated dystrophin gene, excluding disrupting mutations.
Currently, there are two trials ongoing by Sarepta Therapeutics and Prosensa, utilizing two different chemistries: the 2’O-methyl phosphorothioate backbone (2OMePS, named PRO051/Drisapersen initiated by Prosensa/GSK) and the morpholino backbone (PMO, named Eteplirsen initiated by AVI Biopharma, now Sarepta Therapeutics).
Both of these trials primarily target the skipping of exon 51. One of the main lessons learned from these trials over the last 7 years is that there is a strong correlation between dose-per-body-weight, exon-skipping efficiency, and dystrophin production. It appears that doses of 30 to 50mg/kg are needed to obtain favorable increases in dystrophin levels.
Unfortunately, Drisapersen, even at 6mg/kg, has been associated with kidney toxicity, whereas Eteplirsen (morpholino backbone) has not been associated with any toxicity even at higher dose levels.
Efforts are underway to fund other trials that target different mutations (other than exon 51). This approach aims to increase knowledge of exon-skipping processes, find better ways of administering the drug (currently requiring ongoing repeated intravenous administration), and, more importantly, work toward obtaining FDA class approval for the drug, making it applicable to all mutations regardless of their location.
Gene Therapy: Micro-Dystrophin 4900 bases
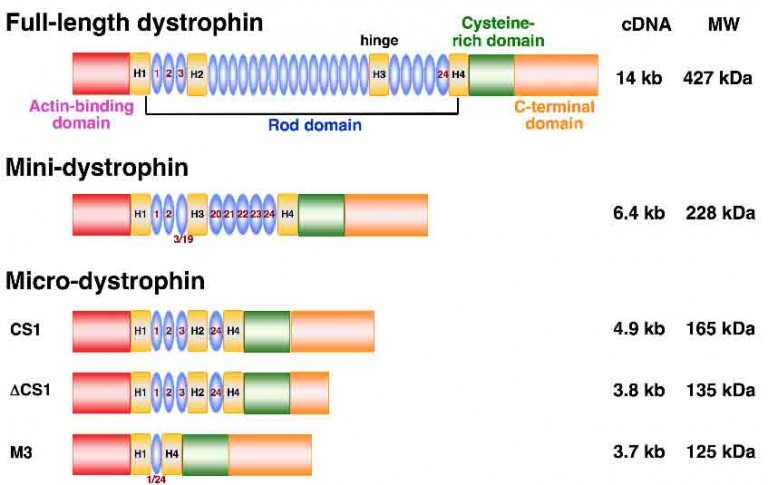
In skeletal and cardiac muscles, dystrophin is part of a group of proteins (a protein complex) that work together to strengthen muscle fibers and protect them from injury as muscles contract and relax. The dystrophin complex acts as an anchor, connecting each muscle cell’s structural framework (cytoskeleton) with the lattice of proteins and other molecules outside the cell (extracellular matrix).
Domains of the dystrophin molecule:
The two ends: the N- and C- terminal domains are the part of the anchor that connects the two ends of the dystrophin protein to the cytoskeleton and the extracellular matrix. The N terminus contains the primary actin-binding site, whereas the C terminus contains the β-dystroglycan, dystrobrevin, and syntrophin-binding sites.
Central rod: the central rod acts like a spring between the two ends (N and C terminal domains) protecting muscle fiber from injury as muscles contract and relax. N- and C-terminal domains are connected by 24 spectrin-like repeats.
The N and C terminal domains have to be there for dystrophin to function. The central rod (24 spectrin-like repeats), however, can be shortened to create a smaller dystrophin that is functional but not as effective (not as flexible) as the full-length dystrophin. The shorter the central rod gets, the less flexible the dystrophin gets, and therefore the possibility of muscle fiber being damaged increases.
Micro dystrophin is 4.9 kb long, meaning the central rod is about 9.1 KB shorter than the full-length dystrophin. Micro-dystrophin is therefore much less flexible and effective in keeping muscle fiber from being damaged; however, it is better than no dystrophin at all as it provides some protection.
AAV.Mini-Dystrophin Overlapping Vectors 6400 bases

Mini dystrophin is about 6400 bases and therefor more flexible and effective in protecting muscle fiber from being damaged. However, due to its size it cannot be delivered with an adeno-associated virus (AAV). To overcome this obstacle, two separate AAVs (hence duel vectors) are used and the gene is split into two parts–a front and a back part. The gene is then delivered into the muscle cells via the two AAVs. The two parts recombine inside the muscle cell to form the entire gene and then the protein.